Takeda Announces U.S. FDA Approval of Supplemental New Drug Application (sNDA) for ICLUSIG® (ponatinib) in Adult Patients with Newly Diagnosed Ph+ ALL
March 19, 2024
Discovering and developing innovative therapies to deliver potentially transformative treatments
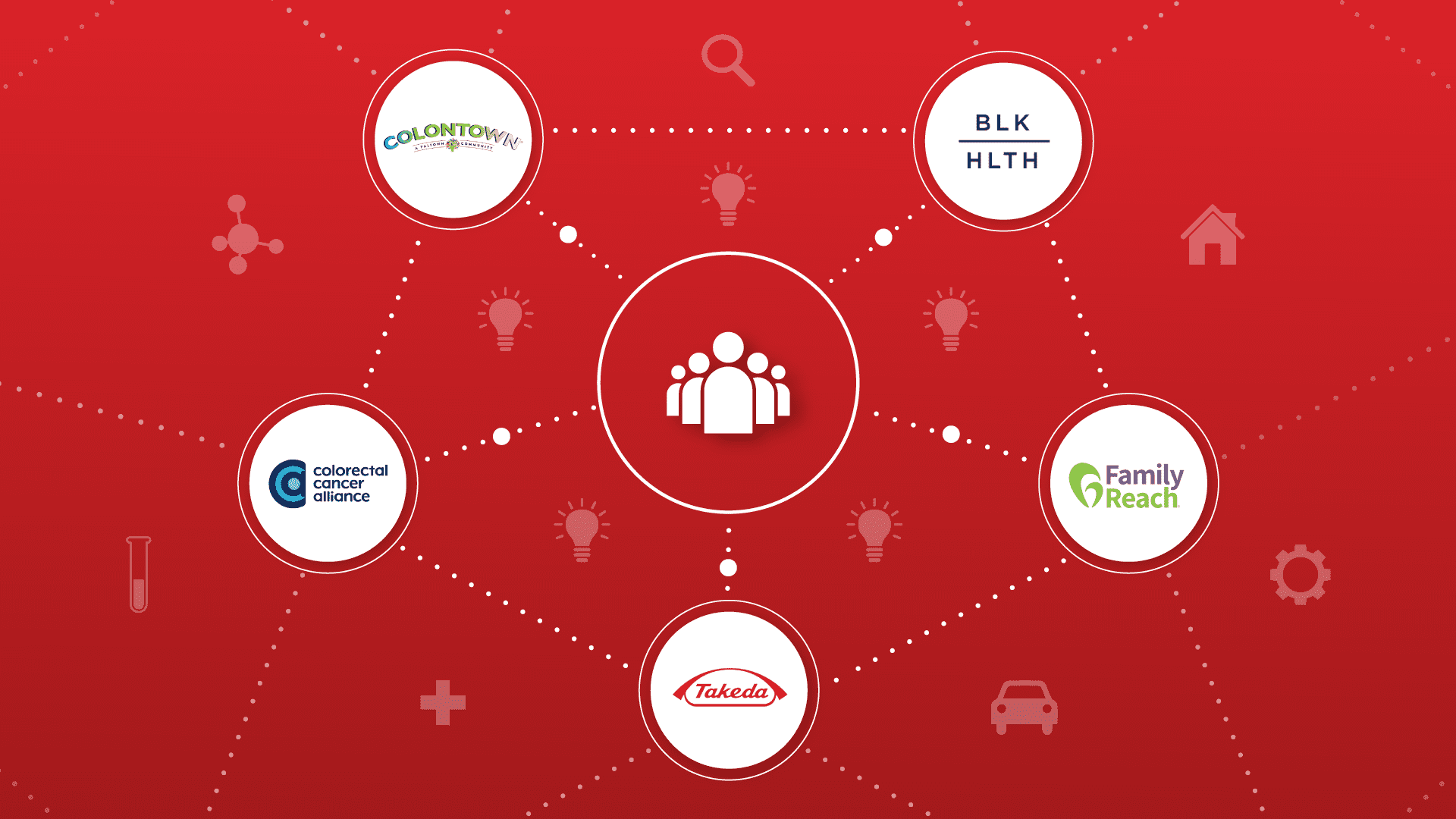
Beyond early detection, how do we bridge the gap in metastatic colorectal cancer care. Learn about Takeda’s partnerships for patient support.